sbiofit
Perform nonlinear least-squares regression
Optimization Toolbox™ and Global Optimization Toolbox are recommended for this function.
Syntax
Description
fitResults = sbiofit(sm,grpData,ResponseMap,estiminfo)sm using
nonlinear least-squares regression.
grpData is a groupedData object specifying
the data to fit. ResponseMap defines the mapping between
the model components and response data in grpData.
estimatedInfo is an EstimatedInfo object that
defines the estimated parameters in the model sm.
fitResults is a OptimResults object or
NLINResults object or a vector
of these objects.
sbiofit uses the first available estimation function among the following:
lsqnonlin (Optimization Toolbox), nlinfit (Statistics and Machine Learning Toolbox), or fminsearch.
By default, each group in grpData is fit separately, resulting in
group-specific parameter estimates. If the model contains active doses and
variants, they are applied before the simulation.
fitResults = sbiofit(sm,grpData,ResponseMap,estiminfo,dosing)dosing instead of using the active doses of the model
sm if there is any.
fitResults = sbiofit(sm,grpData,ResponseMap,estiminfo,dosing,functionName)functionName. If
the specified function is unavailable, a warning is issued and the first
available default function is used.
fitResults = sbiofit(sm,grpData,ResponseMap,estiminfo,dosing,functionName,options)options for the
function functionName.
fitResults = sbiofit(sm,grpData,ResponseMap,estiminfo,dosing,functionName,options,variants)variants instead of
using any active variants of the model.
fitResults = sbiofit(_,Name=Value)
[ also returns a vector of fitResults,simdata]
= sbiofit(_)SimData objects
simdata using any of the input arguments in the
previous syntaxes.
Note
sbiofit simulates the model using a SimFunction object,
which automatically accelerates simulations by default. Hence it is not
necessary to run sbioaccelerate before you
call sbiofit.
Examples
Background
This example shows how to fit an individual's PK profile data to one-compartment model and estimate pharmacokinetic parameters.
Suppose you have drug plasma concentration data from an individual and want to estimate the volume of the central compartment and the clearance. Assume the drug concentration versus the time profile follows the monoexponential decline , where is the drug concentration at time t, is the initial concentration, and is the elimination rate constant that depends on the clearance and volume of the central compartment .
The synthetic data in this example was generated using the following model, parameters, and dose:
One-compartment model with bolus dosing and first-order elimination
Volume of the central compartment (
Central) = 1.70 literClearance parameter (
Cl_Central) = 0.55 liter/hourConstant error model
Bolus dose of 10 mg
Load Data and Visualize
The data is stored as a table with variables Time and Conc that represent the time course of the plasma concentration of an individual after an intravenous bolus administration measured at 13 different time points. The variable units for Time and Conc are hour and milligram/liter, respectively.
load("data15.mat") plot(data.Time,data.Conc,"b+") xlabel("Time (hour)"); ylabel("Drug Concentration (milligram/liter)");

Convert to groupedData Format
Convert the data set to a groupedData object, which is the required data format for the fitting function sbiofit for later use. A groupedData object also lets you set independent variable and group variable names (if they exist). Set the units of the Time and Conc variables. The units are optional and only required for the UnitConversion feature, which automatically converts matching physical quantities to one consistent unit system.
gData = groupedData(data); gData.Properties.VariableUnits = ["hour","milligram/liter"]; gData.Properties
ans = struct with fields:
Description: ''
UserData: []
DimensionNames: {'Row' 'Variables'}
VariableNames: {'Time' 'Conc'}
VariableTypes: ["double" "double"]
VariableDescriptions: {}
VariableUnits: {'hour' 'milligram/liter'}
VariableContinuity: []
RowNames: {}
CustomProperties: [1×1 matlab.tabular.CustomProperties]
GroupVariableName: ''
IndependentVariableName: 'Time'
groupedData automatically set the name of the IndependentVariableName property to the Time variable of the data.
Construct a One-Compartment Model
Use the built-in PK library to construct a one-compartment model with bolus dosing and first-order elimination where the elimination rate depends on the clearance and volume of the central compartment. Use the configset object to turn on unit conversion.
pkmd = PKModelDesign; pkc1 = addCompartment(pkmd,"Central"); pkc1.DosingType = "Bolus"; pkc1.EliminationType = "linear-clearance"; pkc1.HasResponseVariable = true; model = construct(pkmd); configset = getconfigset(model); configset.CompileOptions.UnitConversion = true;
For details on creating compartmental PK models using the SimBiology® built-in library, see Create Pharmacokinetic Models.
Define Dosing
Define a single bolus dose of 10 milligram given at time = 0. For details on setting up different dosing schedules, see Doses in SimBiology Models.
dose = sbiodose("dose"); dose.TargetName = "Drug_Central"; dose.StartTime = 0; dose.Amount = 10; dose.AmountUnits = "milligram"; dose.TimeUnits = "hour";
Map Response Data to the Corresponding Model Component
The data contains drug concentration data stored in the Conc variable. This data corresponds to the Drug_Central species in the model. Therefore, map the data to Drug_Central as follows.
responseMap = "Drug_Central = Conc";Specify Parameters to Estimate
The parameters to fit in this model are the volume of the central compartment (Central) and the clearance rate (Cl_Central). In this case, specify log-transformation for these biological parameters since they are constrained to be positive. The estimatedInfo object lets you specify parameter transforms, initial values, and parameter bounds if needed.
paramsToEstimate = ["log(Central)","log(Cl_Central)"]; estimatedParams = estimatedInfo(paramsToEstimate,InitialValue=[1 1],Bounds=[1 5;0.5 2]);
Estimate Parameters
Now that you have defined one-compartment model, data to fit, mapped response data, parameters to estimate, and dosing, use sbiofit to estimate parameters. The default estimation function that sbiofit uses will change depending on which toolboxes are available. To see which function was used during fitting, check the EstimationFunction property of the corresponding results object.
fitConst = sbiofit(model,gData,responseMap,estimatedParams,dose);
Display Estimated Parameters and Plot Results
Notice the parameter estimates were not far off from the true values (1.70 and 0.55) that were used to generate the data. You may also try different error models to see if they could further improve the parameter estimates.
fitConst.ParameterEstimates
ans=2×4 table
Name Estimate StandardError Bounds
______________ ________ _____________ __________
{'Central' } 1.6993 0.034821 1 5
{'Cl_Central'} 0.53358 0.01968 0.5 2
s.Labels.XLabel = "Time (hour)"; s.Labels.YLabel = "Concentration (milligram/liter)"; plot(fitConst,AxesStyle=s);

Use Different Error Models
Try three other supported error models (proportional, combination of constant and proportional error models, and exponential).
fitProp = sbiofit(model,gData,responseMap,estimatedParams,dose,... ErrorModel="proportional"); fitExp = sbiofit(model,gData,responseMap,estimatedParams,dose,... ErrorModel="exponential"); fitComb = sbiofit(model,gData,responseMap,estimatedParams,dose,... ErrorModel="combined");
Use Weights Instead of an Error Model
You can specify weights as a numeric matrix, where the number of columns corresponds to the number of responses. Setting all weights to 1 is equivalent to the constant error model.
weightsNumeric = ones(size(gData.Conc)); fitWeightsNumeric = sbiofit(model,gData,responseMap,estimatedParams,dose,Weights=weightsNumeric);
Alternatively, you can use a function handle that accepts a vector of predicted response values and returns a vector of weights. In this example, use a function handle that is equivalent to the proportional error model.
weightsFunction = @(y) 1./y.^2; fitWeightsFunction = sbiofit(model,gData,responseMap,estimatedParams,dose,Weights=weightsFunction);
Display Estimated Parameter Values
Show the estimated parameter values of each model.
allResults = [fitConst,fitWeightsNumeric,fitWeightsFunction,fitProp,fitExp,fitComb]; Estimated_Central = zeros(6,1); Estimated_Cl_Central = zeros(6,1); t2 = table(Estimated_Central,Estimated_Cl_Central); errorModelNames = ["constant error model","equal weights","proportional weights", ... "proportional error model","exponential error model",... "combined error model"]; t2.Properties.RowNames = errorModelNames; for i = 1:height(t2) t2{i,1} = allResults(i).ParameterEstimates.Estimate(1); t2{i,2} = allResults(i).ParameterEstimates.Estimate(2); end t2
t2=6×2 table
Estimated_Central Estimated_Cl_Central
_________________ ____________________
constant error model 1.6993 0.53358
equal weights 1.6993 0.53358
proportional weights 1.9045 0.51734
proportional error model 1.8777 0.51147
exponential error model 1.7872 0.51701
combined error model 1.7008 0.53271
Conclusion
This example showed how to estimate PK parameters, namely the volume of the central compartment and clearance parameter of an individual, by fitting the PK profile data to one-compartment model. It showed how to use different error models and numeric weights instead of an error model.
Suppose you have drug plasma concentration data from three individuals that you want
to use to estimate corresponding pharmacokinetic parameters, namely the volume of
central and peripheral compartment (Central,
Peripheral), the clearance rate (Cl_Central),
and intercompartmental clearance (Q12). Assume the drug concentration
versus the time profile follows the biexponential decline , where Ct is the drug
concentration at time t, and a and
b are slopes for corresponding exponential declines.
The synthetic data set contains drug plasma concentration data measured in both central and peripheral compartments. The data was generated using a two-compartment model with an infusion dose and first-order elimination. These parameters were used for each individual.
Central | Peripheral | Q12 | Cl_Central | |
|---|---|---|---|---|
| Individual 1 | 1.90 | 0.68 | 0.24 | 0.57 |
| Individual 2 | 2.10 | 6.05 | 0.36 | 0.95 |
| Individual 3 | 1.70 | 4.21 | 0.46 | 0.95 |
The data is stored as a table with variables ID, Time, CentralConc, and PeripheralConc. It represents the time course of plasma concentrations measured at eight different time points for both central and peripheral compartments after an infusion dose.
load('data10_32R.mat')
Convert the data set to a groupedData object which is the required
data format for the fitting function sbiofit for later use. A
groupedData object also lets you set independent variable and
group variable names (if they exist). Set the units of the ID,
Time, CentralConc, and
PeripheralConc variables. The units are optional and only
required for the UnitConversion feature, which
automatically converts matching physical quantities to one consistent unit
system.
gData = groupedData(data);
gData.Properties.VariableUnits = {'','hour','milligram/liter','milligram/liter'};
gData.Properties
ans =
struct with fields:
Description: ''
UserData: []
DimensionNames: {'Row' 'Variables'}
VariableNames: {'ID' 'Time' 'CentralConc' 'PeripheralConc'}
VariableTypes: ["double" "double" "double" "double"]
VariableDescriptions: {}
VariableUnits: {'' 'hour' 'milligram/liter' 'milligram/liter'}
VariableContinuity: []
RowNames: {}
CustomProperties: [1×1 matlab.tabular.CustomProperties]
GroupVariableName: 'ID'
IndependentVariableName: 'Time'
Create a trellis plot that shows the PK profiles of three individuals.
sbiotrellis(gData,'ID','Time',{'CentralConc','PeripheralConc'},... 'Marker','+','LineStyle','none');

Use the built-in PK library to construct a two-compartment model with infusion dosing and first-order elimination where the elimination rate depends on the clearance and volume of the central compartment. Use the configset object to turn on unit conversion.
pkmd = PKModelDesign; pkc1 = addCompartment(pkmd,'Central'); pkc1.DosingType = 'Infusion'; pkc1.EliminationType = 'linear-clearance'; pkc1.HasResponseVariable = true; pkc2 = addCompartment(pkmd,'Peripheral'); model = construct(pkmd); configset = getconfigset(model); configset.CompileOptions.UnitConversion = true;
Assume every individual receives an infusion dose at time = 0, with a total infusion amount of 100 mg at a rate of 50 mg/hour. For details on setting up different dosing strategies, see Doses in SimBiology Models.
dose = sbiodose('dose','TargetName','Drug_Central'); dose.StartTime = 0; dose.Amount = 100; dose.Rate = 50; dose.AmountUnits = 'milligram'; dose.TimeUnits = 'hour'; dose.RateUnits = 'milligram/hour';
The data contains measured plasma concentrations in the central and peripheral compartments. Map these variables to the appropriate model species, which are Drug_Central and Drug_Peripheral.
responseMap = {'Drug_Central = CentralConc','Drug_Peripheral = PeripheralConc'};
The parameters to estimate in this model are the volumes of central and peripheral compartments (Central and Peripheral), intercompartmental clearance Q12, and clearance rate Cl_Central. In this case, specify log-transform for Central and Peripheral since they are constrained to be positive. The estimatedInfo object lets you specify parameter transforms, initial values, and parameter bounds (optional).
paramsToEstimate = {'log(Central)','log(Peripheral)','Q12','Cl_Central'};
estimatedParam = estimatedInfo(paramsToEstimate,'InitialValue',[1 1 1 1]);
Fit the model to all of the data pooled together, that is, estimate one set of parameters for all individuals. The default estimation method that sbiofit uses will change depending on which toolboxes are available. To see which estimation function sbiofit used for the fitting, check the EstimationFunction property of the corresponding results object.
pooledFit = sbiofit(model,gData,responseMap,estimatedParam,dose,'Pooled',true)
pooledFit =
OptimResults with properties:
ExitFlag: 3
Output: [1×1 struct]
GroupName: []
Beta: [4×3 table]
ParameterEstimates: [4×3 table]
J: [24×4×2 double]
COVB: [4×4 double]
CovarianceMatrix: [4×4 double]
R: [24×2 double]
MSE: 6.6220
SSE: 291.3688
Weights: []
LogLikelihood: -111.3904
AIC: 230.7808
BIC: 238.2656
DFE: 44
DependentFiles: {1×3 cell}
Data: [24×4 groupedData]
EstimatedParameterNames: {'Central' 'Peripheral' 'Q12' 'Cl_Central'}
ErrorModelInfo: [1×3 table]
EstimationFunction: 'lsqnonlin'
Plot the fitted results versus the original data. Although three separate plots were generated, the data was fitted using the same set of parameters (that is, all three individuals had the same fitted line).
plot(pooledFit);

Estimate one set of parameters for each individual and see if there is any improvement in the parameter estimates. In this example, since there are three individuals, three sets of parameters are estimated.
unpooledFit = sbiofit(model,gData,responseMap,estimatedParam,dose,'Pooled',false);
Plot the fitted results versus the original data. Each individual was fitted differently (that is, each fitted line is unique to each individual) and each line appeared to fit well to individual data.
plot(unpooledFit);

Display the fitted results of the first individual. The MSE was lower than that of the pooled fit. This is also true for the other two individuals.
unpooledFit(1)
ans =
OptimResults with properties:
ExitFlag: 3
Output: [1×1 struct]
GroupName: 1
Beta: [4×3 table]
ParameterEstimates: [4×3 table]
J: [8×4×2 double]
COVB: [4×4 double]
CovarianceMatrix: [4×4 double]
R: [8×2 double]
MSE: 2.1380
SSE: 25.6559
Weights: []
LogLikelihood: -26.4805
AIC: 60.9610
BIC: 64.0514
DFE: 12
DependentFiles: {1×3 cell}
Data: [8×4 groupedData]
EstimatedParameterNames: {'Central' 'Peripheral' 'Q12' 'Cl_Central'}
ErrorModelInfo: [1×3 table]
EstimationFunction: 'lsqnonlin'
Generate a plot of the residuals over time to compare the pooled and unpooled fit results. The figure indicates unpooled fit residuals are smaller than those of pooled fit as expected. In addition to comparing residuals, other rigorous criteria can be used to compare the fitted results.
t = [gData.Time;gData.Time]; res_pooled = vertcat(pooledFit.R); res_pooled = res_pooled(:); res_unpooled = vertcat(unpooledFit.R); res_unpooled = res_unpooled(:); plot(t,res_pooled,'o','MarkerFaceColor','w','markerEdgeColor','b') hold on plot(t,res_unpooled,'o','MarkerFaceColor','b','markerEdgeColor','b') refl = refline(0,0); % A reference line representing a zero residual title('Residuals versus Time'); xlabel('Time'); ylabel('Residuals'); legend({'Pooled','Unpooled'});

This example showed how to perform pooled and unpooled estimations using sbiofit. As illustrated, the unpooled fit accounts for variations due to the specific subjects in the study, and, in this case, the model fits better to the data. However, the pooled fit returns population-wide parameters. If you want to estimate population-wide parameters while considering individual variations, use sbiofitmixed.
This example shows how to estimate category-specific (such as young versus old, male versus female), individual-specific, and population-wide parameters using PK profile data from multiple individuals.
Background
Suppose you have drug plasma concentration data from 30 individuals and want to estimate pharmacokinetic parameters, namely the volumes of central and peripheral compartment, the clearance, and intercompartmental clearance. Assume the drug concentration versus the time profile follows the biexponential decline , where is the drug concentration at time t, and and are slopes for corresponding exponential declines.
Load Data
This synthetic data contains the time course of plasma concentrations of 30 individuals after a bolus dose (100 mg) measured at different times for both central and peripheral compartments. It also contains categorical variables, namely Sex and Age.
clear
load('sd5_302RAgeSex.mat')Convert to groupedData Format
Convert the data set to a groupedData object, which is the required data format for the fitting function sbiofit. A groupedData object also allows you set independent variable and group variable names (if they exist). Set the units of the ID, Time, CentralConc, PeripheralConc, Age, and Sex variables. The units are optional and only required for the UnitConversion feature, which automatically converts matching physical quantities to one consistent unit system.
gData = groupedData(data);
gData.Properties.VariableUnits = {'','hour','milligram/liter','milligram/liter','',''};
gData.Propertiesans = struct with fields:
Description: ''
UserData: []
DimensionNames: {'Row' 'Variables'}
VariableNames: {'ID' 'Time' 'CentralConc' 'PeripheralConc' 'Sex' 'Age'}
VariableTypes: ["double" "double" "double" "double" "categorical" "categorical"]
VariableDescriptions: {}
VariableUnits: {'' 'hour' 'milligram/liter' 'milligram/liter' '' ''}
VariableContinuity: []
RowNames: {}
CustomProperties: [1×1 matlab.tabular.CustomProperties]
GroupVariableName: 'ID'
IndependentVariableName: 'Time'
The IndependentVariableName and GroupVariableName properties have been automatically set to the Time and ID variables of the data.
Visualize Data
Display the response data for each individual.
t = sbiotrellis(gData,'ID','Time',{'CentralConc','PeripheralConc'},... 'Marker','+','LineStyle','none'); % Resize the figure. t.hFig.Position(:) = [100 100 1280 800];

Set Up a Two-Compartment Model
Use the built-in PK library to construct a two-compartment model with infusion dosing and first-order elimination where the elimination rate depends on the clearance and volume of the central compartment. Use the configset object to turn on unit conversion.
pkmd = PKModelDesign; pkc1 = addCompartment(pkmd,'Central'); pkc1.DosingType = 'Bolus'; pkc1.EliminationType = 'linear-clearance'; pkc1.HasResponseVariable = true; pkc2 = addCompartment(pkmd,'Peripheral'); model = construct(pkmd); configset = getconfigset(model); configset.CompileOptions.UnitConversion = true;
For details on creating compartmental PK models using the SimBiology® built-in library, see Create Pharmacokinetic Models.
Define Dosing
Assume every individual receives a bolus dose of 100 mg at time = 0. For details on setting up different dosing strategies, see Doses in SimBiology Models.
dose = sbiodose('dose','TargetName','Drug_Central'); dose.StartTime = 0; dose.Amount = 100; dose.AmountUnits = 'milligram'; dose.TimeUnits = 'hour';
Map the Response Data to Corresponding Model Components
The data contains measured plasma concentration in the central and peripheral compartments. Map these variables to the appropriate model components, which are Drug_Central and Drug_Peripheral.
responseMap = {'Drug_Central = CentralConc','Drug_Peripheral = PeripheralConc'};Specify Parameters to Estimate
Specify the volumes of central and peripheral compartments Central and Peripheral, intercompartmental clearance Q12, and clearance Cl_Central as parameters to estimate. The estimatedInfo object lets you optionally specify parameter transforms, initial values, and parameter bounds. Since both Central and Peripheral are constrained to be positive, specify a log-transform for each parameter.
paramsToEstimate = {'log(Central)', 'log(Peripheral)', 'Q12', 'Cl_Central'};
estimatedParam = estimatedInfo(paramsToEstimate,'InitialValue',[1 1 1 1]);Estimate Individual-Specific Parameters
Estimate one set of parameters for each individual by setting the 'Pooled' name-value pair argument to false.
unpooledFit = sbiofit(model,gData,responseMap,estimatedParam,dose,'Pooled',false);Display Results
Plot the fitted results versus the original data for each individual (group).
plot(unpooledFit);

For an unpooled fit, sbiofit always returns one results object for each individual.
Examine Parameter Estimates for Category Dependencies
Explore the unpooled estimates to see if there is any category-specific parameters, that is, if some parameters are related to one or more categories. If there are any category dependencies, it might be possible to reduce the number of degrees of freedom by estimating just category-specific values for those parameters.
First extract the ID and category values for each ID
catParamValues = unique(gData(:,{'ID','Sex','Age'}));Add variables to the table containing each parameter's estimate.
allParamValues = vertcat(unpooledFit.ParameterEstimates); catParamValues.Central = allParamValues.Estimate(strcmp(allParamValues.Name, 'Central')); catParamValues.Peripheral = allParamValues.Estimate(strcmp(allParamValues.Name, 'Peripheral')); catParamValues.Q12 = allParamValues.Estimate(strcmp(allParamValues.Name, 'Q12')); catParamValues.Cl_Central = allParamValues.Estimate(strcmp(allParamValues.Name, 'Cl_Central'));
Plot estimates of each parameter for each category. gscatter requires Statistics and Machine Learning Toolbox™. If you do not have it, use other alternative plotting functions such as plot.
h = figure; ylabels = ["Central","Peripheral","Q12","Cl\_Central"]; plotNumber = 1; for i = 1:4 thisParam = estimatedParam(i).Name; % Plot for Sex category subplot(4,2,plotNumber); plotNumber = plotNumber + 1; gscatter(double(catParamValues.Sex), catParamValues.(thisParam), catParamValues.Sex); ax = gca; ax.XTick = []; ylabel(ylabels(i)); legend('Location','bestoutside') % Plot for Age category subplot(4,2,plotNumber); plotNumber = plotNumber + 1; gscatter(double(catParamValues.Age), catParamValues.(thisParam), catParamValues.Age); ax = gca; ax.XTick = []; ylabel(ylabels(i)); legend('Location','bestoutside') end % Resize the figure. h.Position(:) = [100 100 1280 800];

Based on the plot, it seems that young individuals tend to have higher volumes of central and peripheral compartments (Central, Peripheral) than old individuals (that is, the volumes seem to be age-specific). In addition, males tend to have lower clearance rates (Cl_Central) than females but the opposite for the Q12 parameter (that is, the clearance and Q12 seem to be sex-specific).
Estimate Category-Specific Parameters
Use the 'CategoryVariableName' property of the estimatedInfo object to specify which category to use during fitting. Use 'Sex' as the group to fit for the clearance Cl_Central and Q12 parameters. Use 'Age' as the group to fit for the Central and Peripheral parameters.
estimatedParam(1).CategoryVariableName = 'Age'; estimatedParam(2).CategoryVariableName = 'Age'; estimatedParam(3).CategoryVariableName = 'Sex'; estimatedParam(4).CategoryVariableName = 'Sex'; categoryFit = sbiofit(model,gData,responseMap,estimatedParam,dose)
categoryFit =
OptimResults with properties:
ExitFlag: 3
Output: [1×1 struct]
GroupName: []
Beta: [8×5 table]
ParameterEstimates: [120×6 table]
J: [240×8×2 double]
COVB: [8×8 double]
CovarianceMatrix: [8×8 double]
R: [240×2 double]
MSE: 0.4362
SSE: 205.8690
Weights: []
LogLikelihood: -477.9195
AIC: 971.8390
BIC: 1.0052e+03
DFE: 472
DependentFiles: {1×3 cell}
Data: [240×6 groupedData]
EstimatedParameterNames: {'Central' 'Peripheral' 'Q12' 'Cl_Central'}
ErrorModelInfo: [1×3 table]
EstimationFunction: 'lsqnonlin'
When fitting by category (or group), sbiofit always returns one results object, not one for each category level. This is because both male and female individuals are considered to be part of the same optimization using the same error model and error parameters, similarly for the young and old individuals.
Plot Results
Plot the category-specific estimated results.
plot(categoryFit);

For the Cl_Central and Q12 parameters, all males had the same estimates, and similarly for the females. For the Central and Peripheral parameters, all young individuals had the same estimates, and similarly for the old individuals.
Estimate Population-Wide Parameters
To better compare the results, fit the model to all of the data pooled together, that is, estimate one set of parameters for all individuals by setting the 'Pooled' name-value pair argument to true. The warning message tells you that this option will ignore any category-specific information (if they exist).
pooledFit = sbiofit(model,gData,responseMap,estimatedParam,dose,'Pooled',true);Warning: CategoryVariableName property of the estimatedInfo object is ignored when using the 'Pooled' option.
Plot Results
Plot the fitted results versus the original data. Although a separate plot was generated for each individual, the data was fitted using the same set of parameters (that is, all individuals had the same fitted line).
plot(pooledFit);

Compare Residuals
Compare residuals of CentralConc and PeripheralConc responses for each fit.
t = gData.Time;
allResid(:,:,1) = pooledFit.R;
allResid(:,:,2) = categoryFit.R;
allResid(:,:,3) = vertcat(unpooledFit.R);
h = figure;
responseList = {'CentralConc', 'PeripheralConc'};
for i = 1:2
subplot(2,1,i);
oneResid = squeeze(allResid(:,i,:));
plot(t,oneResid,'o');
refline(0,0); % A reference line representing a zero residual
title(sprintf('Residuals (%s)', responseList{i}));
xlabel('Time');
ylabel('Residuals');
legend({'Pooled','Category-Specific','Unpooled'});
end
% Resize the figure.
h.Position(:) = [100 100 1280 800];
As shown in the plot, the unpooled fit produced the best fit to the data as it fit the data to each individual. This was expected since it used the most number of degrees of freedom. The category-fit reduced the number of degrees of freedom by fitting the data to two categories (sex and age). As a result, the residuals were larger than the unpooled fit, but still smaller than the population-fit, which estimated just one set of parameters for all individuals. The category-fit might be a good compromise between the unpooled and pooled fitting provided that any hierarchical model exists within your data.
This example uses the yeast heterotrimeric G protein model and experimental data reported by [1]. For details about the model, see the Background section in Parameter Scanning, Parameter Estimation, and Sensitivity Analysis in the Yeast Heterotrimeric G Protein Cycle.
Load the G protein model.
sbioloadproject gproteinStore the experimental data containing the time course for the fraction of active G protein.
time = [0 10 30 60 110 210 300 450 600]'; GaFracExpt = [0 0.35 0.4 0.36 0.39 0.33 0.24 0.17 0.2]';
Create a groupedData object based on the experimental data.
tbl = table(time,GaFracExpt); grpData = groupedData(tbl);
Map the appropriate model component to the experimental data. In other words, indicate which species in the model corresponds to which response variable in the data. In this example, map the model parameter GaFrac to the experimental data variable GaFracExpt from grpData.
responseMap = 'GaFrac = GaFracExpt';Use an estimatedInfo object to define the model parameter kGd as a parameter to be estimated.
estimatedParam = estimatedInfo('kGd');Perform the parameter estimation.
fitResult = sbiofit(m1,grpData,responseMap,estimatedParam);
View the estimated parameter value of kGd.
fitResult.ParameterEstimates
ans=1×3 table
Name Estimate StandardError
_______ ________ _____________
{'kGd'} 0.11307 3.4439e-05
Suppose you want to plot the model simulation results using the estimated parameter value. You can either rerun the sbiofit function and specify to return the optional second output argument, which contains simulation results, or use the fitted method to retrieve the results without rerunning sbiofit.
[yfit,paramEstim] = fitted(fitResult);
Plot the simulation results.
sbioplot(yfit);

This example shows how to estimate the time lag before a bolus dose was administered and the duration of the dose using a one-compartment model.
Load a sample data set.
load lagDurationData.matPlot the data.
plot(data.Time,data.Conc,'x') xlabel('Time (hour)') ylabel('Conc (milligram/liter)')

Convert to groupedData.
gData = groupedData(data);
gData.Properties.VariableUnits = {'hour','milligram/liter'};Create a one-compartment model.
pkmd = PKModelDesign; pkc1 = addCompartment(pkmd,'Central'); pkc1.DosingType = 'Bolus'; pkc1.EliminationType = 'linear-clearance'; pkc1.HasResponseVariable = true; model = construct(pkmd); configset = getconfigset(model); configset.CompileOptions.UnitConversion = true;
Add two parameters that represent the time lag and duration of a dose. The lag parameter specifies the time lag before the dose is administered. The duration parameter specifies the length of time it takes to administer a dose.
lagP = addparameter(model,'lagP'); lagP.ValueUnits = 'hour'; durP = addparameter(model,'durP'); durP.ValueUnits = 'hour';
Create a dose object. Set the LagParameterName and DurationParameterName properties of the dose to the names of the lag and duration parameters, respectively. Set the dose amount to 10 milligram which was the amount used to generate the data.
dose = sbiodose('dose'); dose.TargetName = 'Drug_Central'; dose.StartTime = 0; dose.Amount = 10; dose.AmountUnits = 'milligram'; dose.TimeUnits = 'hour'; dose.LagParameterName = 'lagP'; dose.DurationParameterName = 'durP';
Map the model species to the corresponding data.
responseMap = {'Drug_Central = Conc'};Specify the lag and duration parameters as parameters to estimate. Log-transform the parameters. Initialize them to 2 and set the upper bound and lower bound.
paramsToEstimate = {'log(lagP)','log(durP)'};
estimatedParams = estimatedInfo(paramsToEstimate,'InitialValue',2,'Bounds',[1 5]);Perform parameter estimation.
fitResults = sbiofit(model,gData,responseMap,estimatedParams,dose,'fminsearch')fitResults =
OptimResults with properties:
ExitFlag: 1
Output: [1×1 struct]
GroupName: One group
Beta: [2×4 table]
ParameterEstimates: [2×4 table]
J: [11×2 double]
COVB: [2×2 double]
CovarianceMatrix: [2×2 double]
R: [11×1 double]
MSE: 0.0024
SSE: 0.0213
Weights: []
LogLikelihood: 18.7511
AIC: -33.5023
BIC: -32.7065
DFE: 9
DependentFiles: {1×2 cell}
Data: [11×2 groupedData]
EstimatedParameterNames: {'lagP' 'durP'}
ErrorModelInfo: [1×3 table]
EstimationFunction: 'fminsearch'
Display the result.
fitResults.ParameterEstimates
ans=2×4 table
Name Estimate StandardError Bounds
________ ________ _____________ ______
{'lagP'} 1.986 0.0051568 1 5
{'durP'} 1.527 0.012956 1 5
plot(fitResults)

Import data to use for fitting. It comprises 12 groups of measurements across three responses: Drug, Target, and Complex. The Drug response includes measurements for all 12 groups, whereas the Target and Complex responses have measurements for only 8 and 4 groups, respectively. A group could represent a patient or an experimental or biological condition. Ndrug, Ntarget, and Ncomplex columns represent the constant number of groups that have the measurements for the corresponding responses. Each group has its own dosing amount, represented by the Dose column.
dataTMDD = readtable('tmddData_final.csv');Display the first few rows of the data set.
head(dataTMDD)
ID Time Dose Drug Target Complex Ndrug Ntarget Ncomplex
__ ____ ____ _________ ______ _______ _____ _______ ________
1 0 10 NaN NaN NaN 12 8 4
1 0.5 NaN 1.2131 10.116 0.36602 12 8 4
1 1 NaN 0.74803 9.4694 0.61803 12 8 4
1 2 NaN 0.28601 9.2434 0.84304 12 8 4
1 4 NaN 0.048002 9.6184 0.83204 12 8 4
1 8 NaN 0.0080003 9.6334 0.67203 12 8 4
1 12 NaN 0.0060003 9.4574 0.54002 12 8 4
1 16 NaN 0.0050002 9.7914 0.40502 12 8 4
Convert the data to a groupedData format.
gData = groupedData(dataTMDD); gData.Properties.VariableUnits = ["","hour","nanomole","nanomole/liter","nanomole/liter","nanomole/liter","","",""]; gData.Properties.GroupVariableName = "ID"; gData.Properties.IndependentVariableName = "Time";
Plot the responses: Drug, Target, and Complex.
t = sbiotrellis(gData,"ID","Time","Drug",Marker="+",LineStyle="none"); plot(t,gData,"ID","Time","Target",Marker="o",LineStyle="none"); plot(t,gData,"ID","Time","Complex",Marker="^",LineStyle="none"); t.hFig.Position = [100 100 1280 800]; t.YLabel = "nanomole/liter";

Load the target-mediated drug deposition (TMDD) model. For details about the model, see Scan Dosing Regimens Using SimBiology Model Analyzer App.
sbioloadproject("tmdd_with_TO.sbproj","m1");
The data has 12 groups of measurements, and each group has its own dose amount. Create a corresponding dose object for each group using the dosing information from the data. During the subsequent fitting step, the fit function applies each dose to each group in the same order that the groups appear in the input data. For details on setting up doses, see dosing.
dose = sbiodose("Dose"); dose.TargetName = "Plasma.Drug"; doses = createDoses(gData,"Dose","",dose);
Specify parameters to estimate and their initial values. Also set upper and lower parameter bounds.
paramsToEstimate = estimatedInfo(["log(km)","log(kon)","log(koff)"],InitialValue=0.01,Bounds=[1e-05 1]);
Map the measurement data to the corresponding model components.
responseMap = ["Plasma.Drug = Drug",... "Plasma.Target = Target",... "Plasma.Complex = Complex"];
Define the algorithm options.
options = optimoptions("lsqnonlin");
options.StepTolerance = 1e-08;
options.FunctionTolerance = 1e-08;
options.OptimalityTolerance = 1e-06;
options.MaxIterations = 100;Define the fit problem.
f = fitproblem(FitFunction="sbiofit"); f.Data = gData; f.Estimated = paramsToEstimate; f.Model = m1; f.ResponseMap = responseMap; f.Doses = doses; f.FunctionName = "lsqnonlin"; f.Options = options; f.Pooled = true; f.ProgressPlot = true;
Run the fit with weights set to 1.
f.Weights = [ones(size(gData.Drug)) ones(size(gData.Target)) ones(size(gData.Complex))]; fitEqualWeights = fit(f);

plot(fitEqualWeights);

Plot all groups in one plot for each response.
plot(fitEqualWeights,PlotStyle="one axes");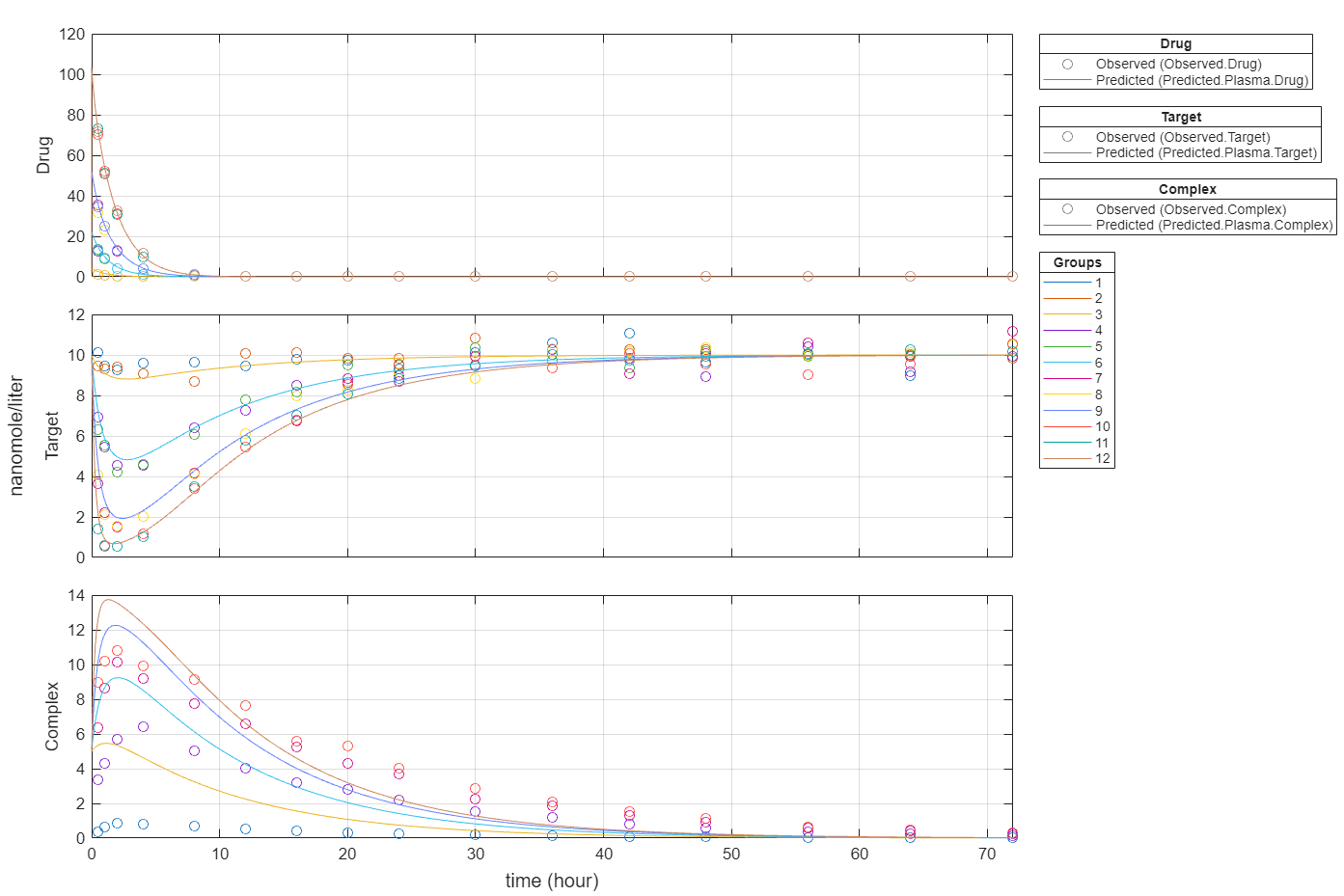
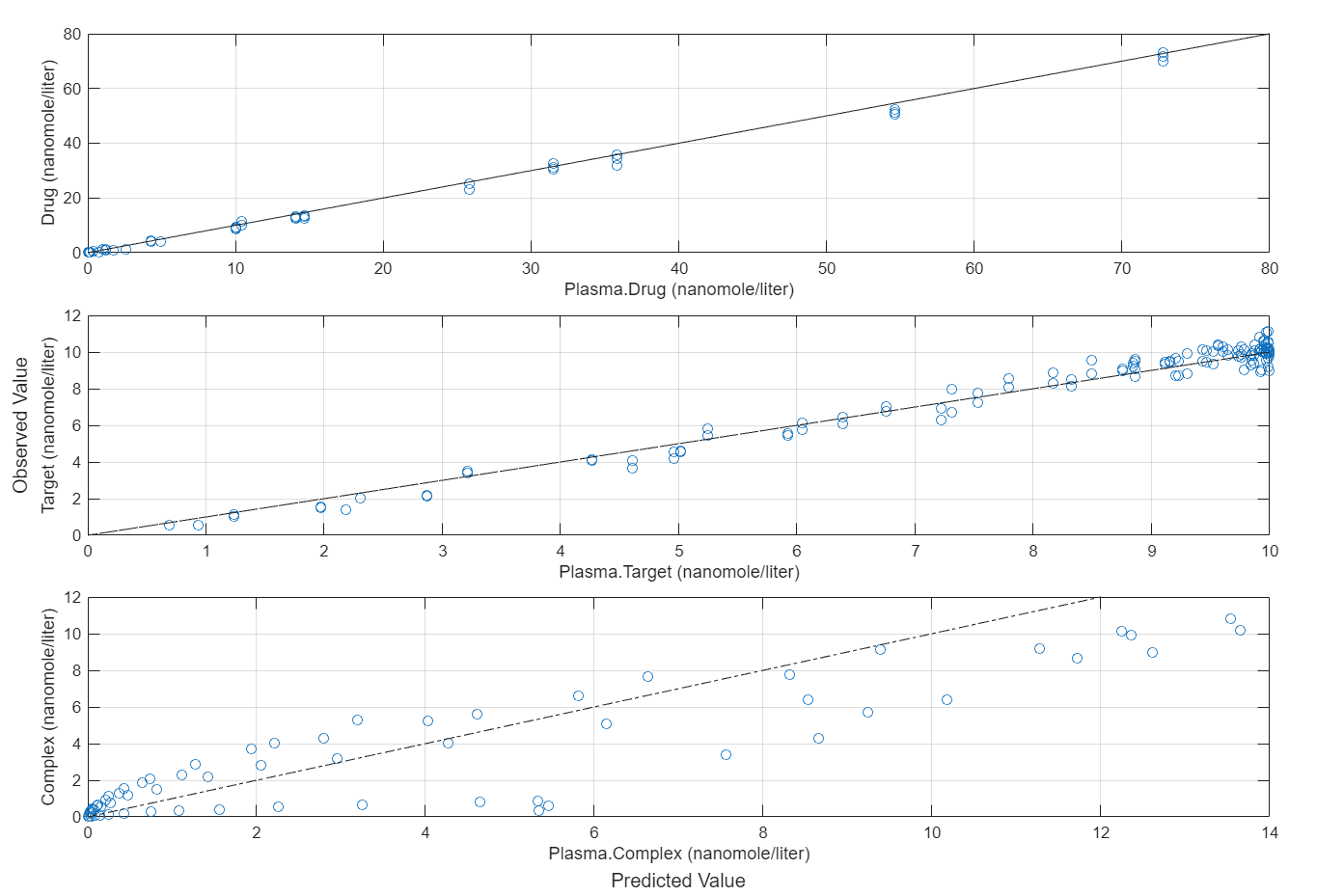
Compare the predictions to the actual data.
plotActualVersusPredicted(fitEqualWeights)
In this data set, the response Drug has 12 groups of measurements, Target has 8 groups of measurements, and Complex has 4 groups of measurements. The varying number of measurements among these responses might stem from the experimental design. To address these differences, you might consider experimenting with weights that are inversely proportional to the number of measurement groups.
f.Weights = [1./gData.Ndrug 1./gData.Ntarget 1./gData.Ncomplex]; fitInverseWeights = fit(f);
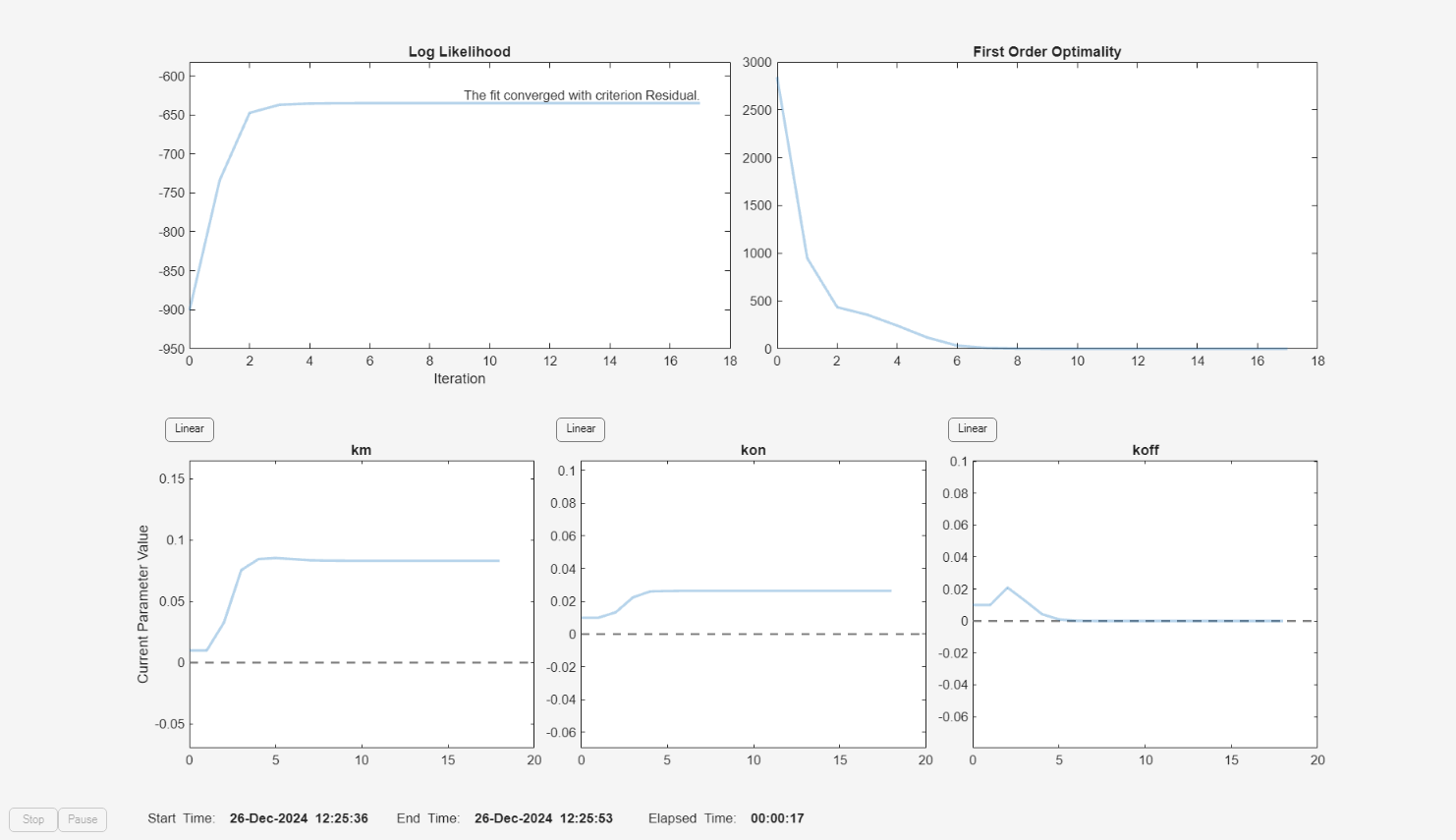
plot(fitInverseWeights);

Plot all groups in one plot for each response.
plot(fitInverseWeights,PlotStyle="one axes");
Compare the predictions to the actual data.
plotActualVersusPredicted(fitInverseWeights)
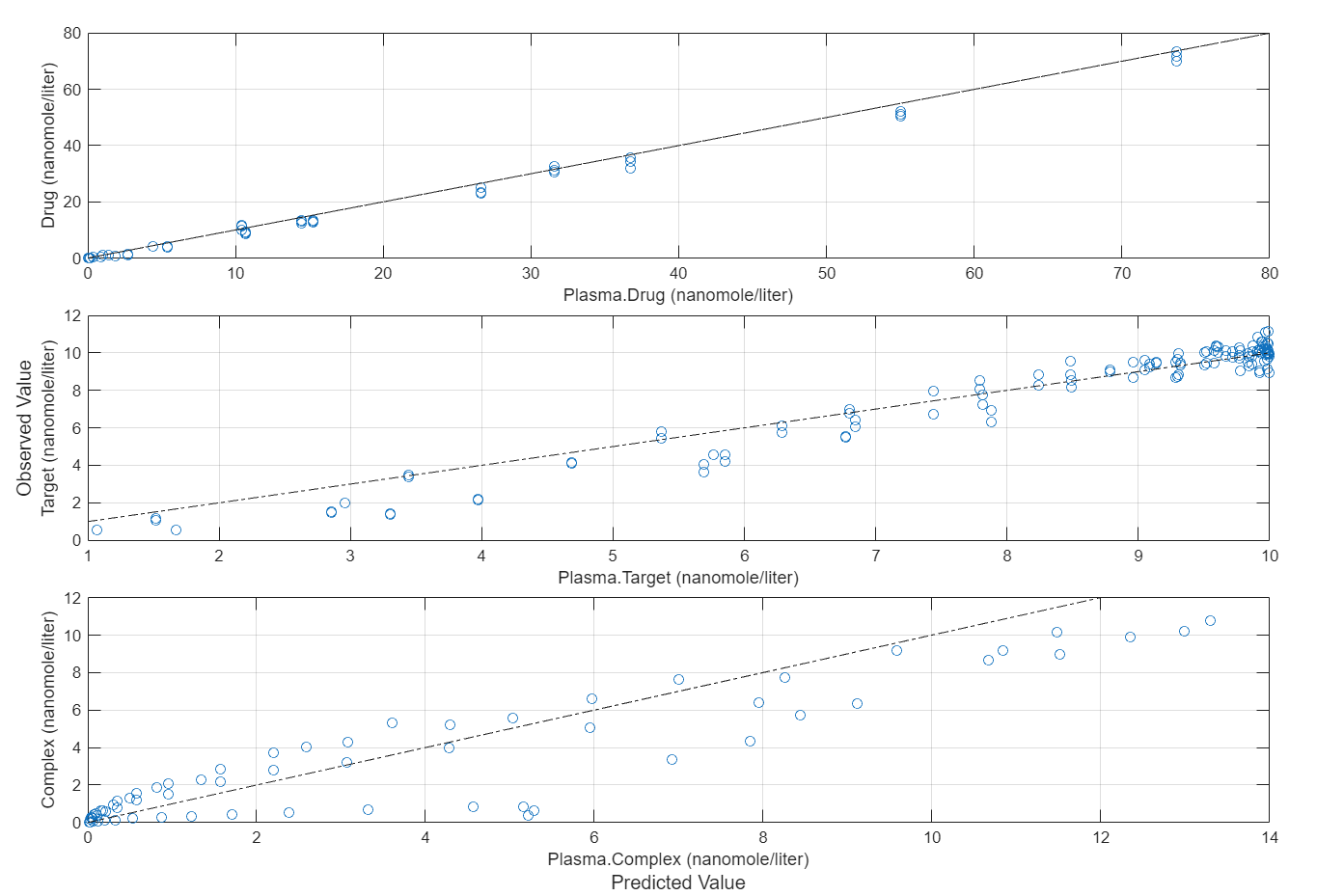
Input Arguments
Name-Value Arguments
Output Arguments
More About
References
[1] Yi, T-M., Kitano, H., and Simon, M. (2003). A quantitative characterization of the yeast heterotrimeric G protein cycle. PNAS. 100, 10764–10769.
[2] Gábor, A., and Banga, J.R. (2015). Robust and efficient parameter estimation in dynamic models of biological systems. BMC Systems Biology. 9:74.
Extended Capabilities
Version History
Introduced in R2014a
See Also
EstimatedInfo object | groupedData object | LeastSquaresResults object | NLINResults object | OptimResults object | sbiofitmixed | nlinfit (Statistics and Machine Learning Toolbox) | fmincon (Optimization Toolbox) | fminunc (Optimization Toolbox) | fminsearch | lsqcurvefit (Optimization Toolbox) | lsqnonlin (Optimization Toolbox) | patternsearch (Global Optimization Toolbox) | ga (Global Optimization Toolbox) | particleswarm (Global Optimization Toolbox)
Topics
- Parameter Scanning, Parameter Estimation, and Sensitivity Analysis in the Yeast Heterotrimeric G Protein Cycle
- What is Nonlinear Regression?
- Fitting Options in SimBiology
- Parameter Transformations
- Maximum Likelihood Estimation
- Fitting Workflow
- Supported Methods for Parameter Estimation in SimBiology
- Sensitivity Analysis in SimBiology
- Progress Plot
- Supported Files and Data Types
- Create Data File with SimBiology Definitions